![]()
Introduction to single-cell RNA-seq I: pre-processing and quality control¶
This R notebook demonstrates the use of the kallisto and bustools programs for pre-processing single-cell RNA-seq data (also available as a Python notebook). It streams in 1 million C. elegans reads, pseudoaligns them, and produces a cells x genes count matrix in about a minute. The notebook then performs some basic QC. It expands on a notebook prepared by Sina Booeshaghi for the Genome Informatics 2019 meeting, where he ran it in under 60 seconds during a 1 minute "lightning talk".
The kallistobus.tools tutorials site has a extensive list of follow-up tutorials and vignettes on single-cell RNA-seq.
1 2 3 4 5 | |
Installing package into ‘/usr/local/lib/R/site-library’
(as ‘lib’ is unspecified)
</head>
<body>
<iframe src="https://www.youtube.com/embed/x-rNofr88BM" width="560" height="315" frameborder="0" allowfullscreen=""></iframe>
</body>
The notebook was written by A. Sina Booeshaghi, Lambda Lu and Lior Pachter. If you use the methods in this notebook for your analysis please cite the following publication, on which it is based:
- Melsted, P., Booeshaghi, A.S. et al. Modular and efficient pre-processing of single-cell RNA-seq. bioRxiv (2019). doi:10.1101/673285
Setup¶
1 2 | |
Install R packages¶
1 | |
Installing packages into ‘/usr/local/lib/R/site-library’
(as ‘lib’ is unspecified)
1 2 3 4 5 6 | |
Install kb-python (includes kallisto and bustools)¶
1 | |
Download required files¶
1 2 3 4 5 6 7 8 | |
Pseudoalignment and counting¶
In this notebook we pseudoalign 1 million C. elegans reads and count UMIs to produce a cells x genes matrix. These are located at XXX and instead of being downloaded, are streamed directly to the Google Colab notebook for quantification. See this blog post for more details on how the streaming works.
The data consists of a subset of reads from GSE126954 described in the paper:
- Packer, J., Zhu, Q. et al. A lineage-resolved molecular atlas of C. elegans embryogenesis at single-cell resolution. Science (2019). doi:10.1126/science.aax1971
Run kallisto and bustools¶
1 2 | |
Basic QC¶
Represent the cells in 2D¶
1 2 | |
1 2 | |
1 | |
- 95372
- 22113
Here cells are in rows and genes are in columns, while usually in single cell analyses, cells are in columns and genes are in rows. Here most "cells" are empty droplets. What if we do PCA now?
1 2 3 | |
1 2 3 | |
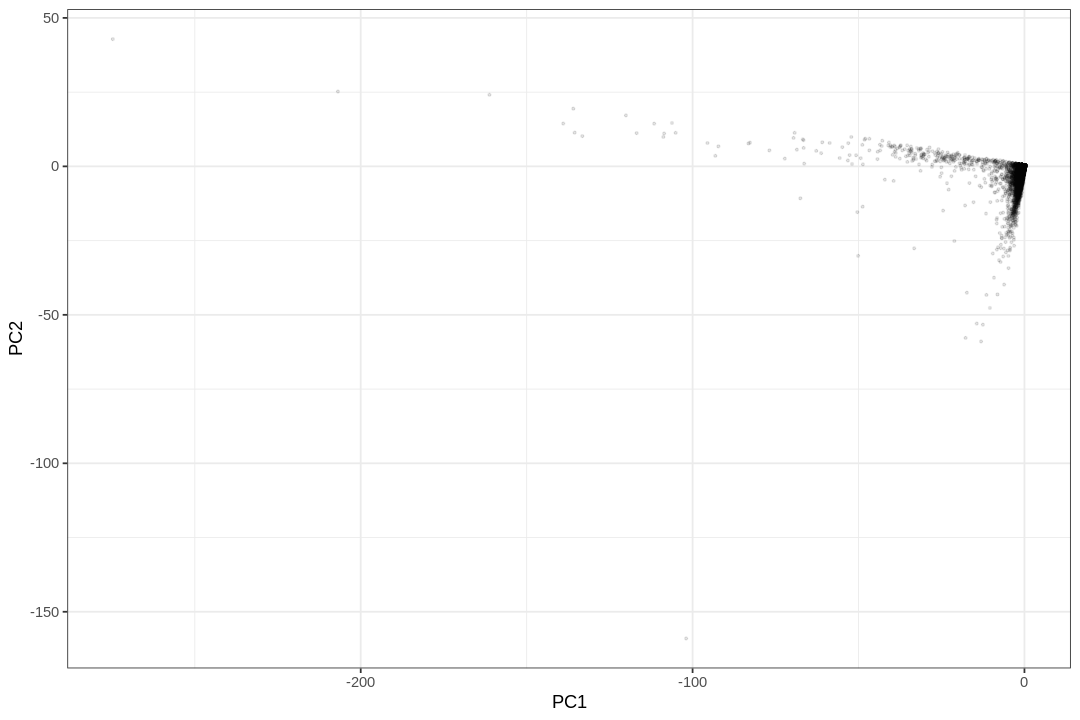
While the PCA plot shows the overall structure of the data, a visualization highlighting the density of points reveals a large number of droplets represented in the lower left corner.
1 2 3 | |
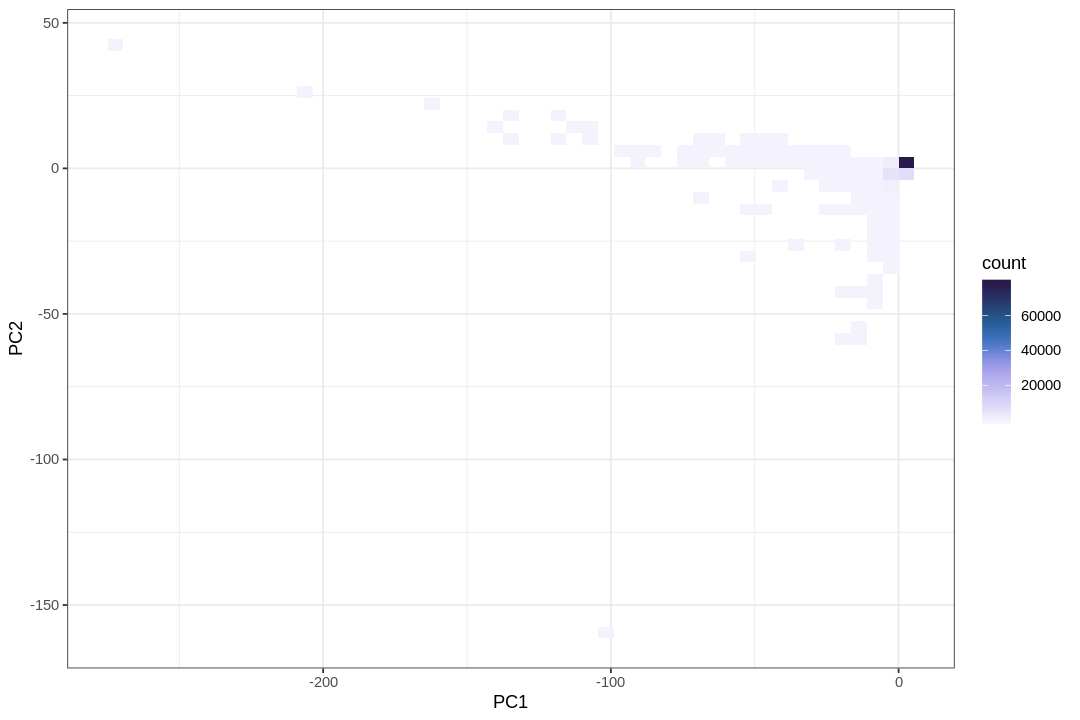
The following plot helps clarify the reason for the concentrated points in the lower-left corner of the PCA plot.
Test for library saturation¶
1 2 | |
1 2 3 4 5 | |
Warning message:
“Transformation introduced infinite values in continuous x-axis”
Warning message:
“Transformation introduced infinite values in continuous y-axis”
Warning message:
“Removed 1018 rows containing non-finite values (stat_bin2d).”
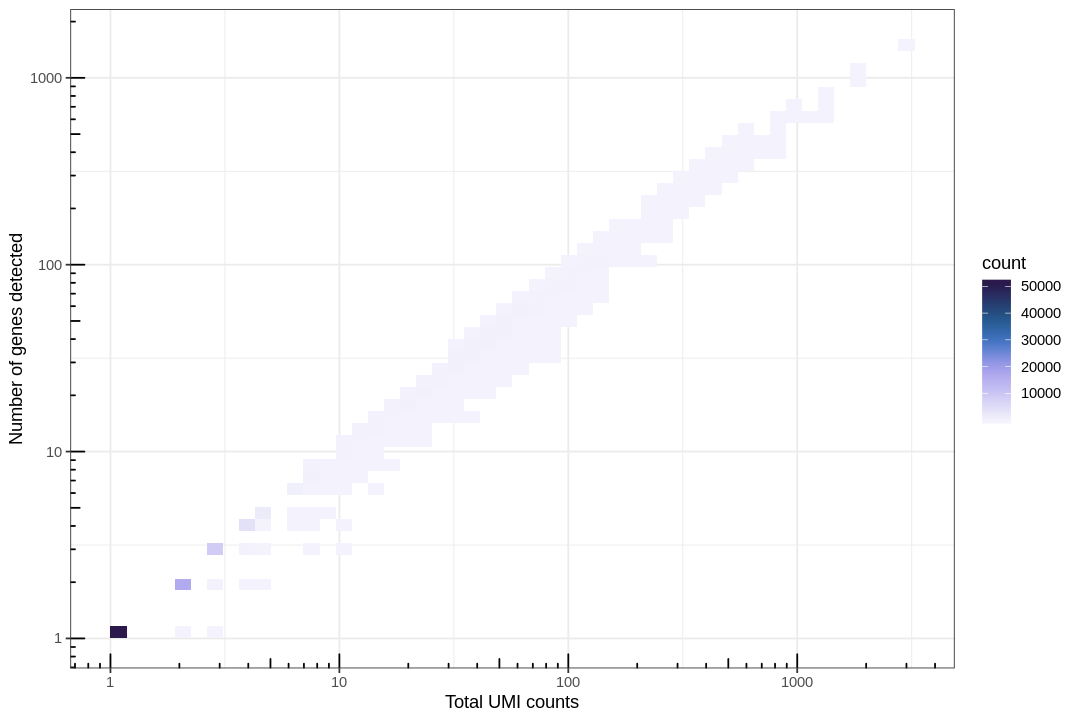
Here we see that there are a large number of near empty droplets. A useful approach to filtering out such data is the "knee plot" shown below.
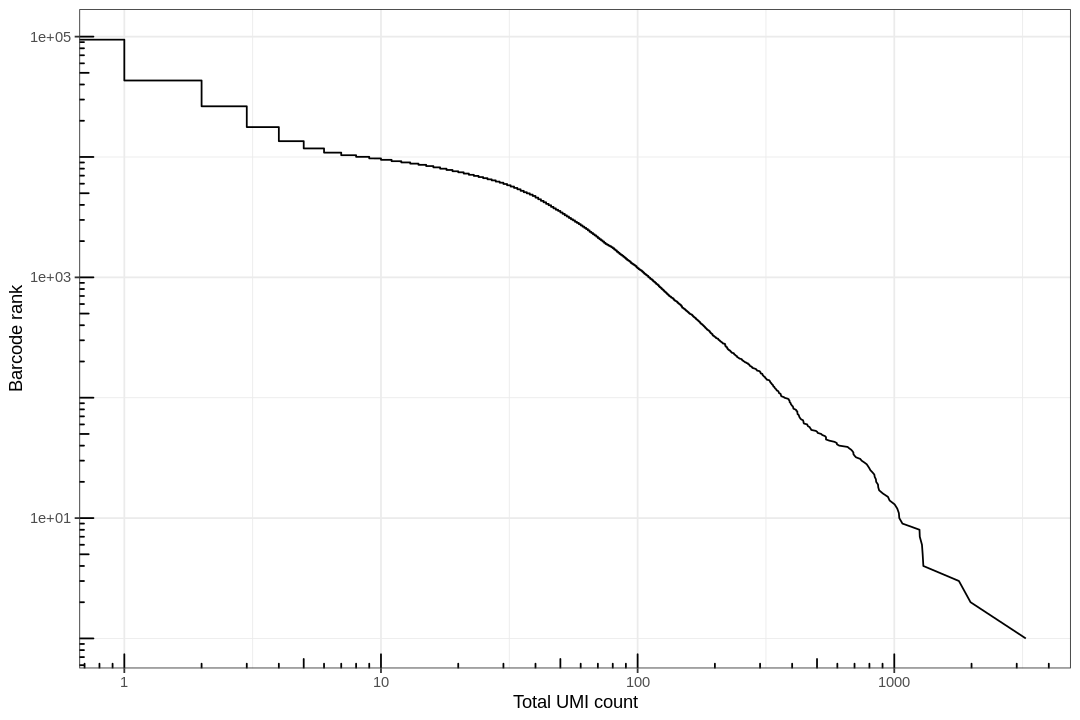
Examine the knee plot¶
The "knee plot" was introduced in the Drop-seq paper: - Macosko et al., Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets, 2015. DOI:10.1016/j.cell.2015.05.002
In this plot cells are ordered by the number of UMI counts associated to them (shown on the x-axis), and the fraction of droplets with at least that number of cells is shown on the y-axis:
1 2 3 4 5 6 7 8 9 10 11 12 | |
Warning message:
“Transformation introduced infinite values in continuous x-axis”
1 2 | |
Exercises¶
- The "knee plot" is sometimes shown with the UMI counts on the y-axis instead of the x-axis, i.e. flipped and rotated 90 degrees. Make the flipped and rotated plot. Is there a reason to prefer one orientation over the other?
1 2 3 4 5 6 7 8 9 10 11 12 | |
For more information on this exercise see Rotating the knee (plot) and related yoga.
Discussion¶
This notebook has demonstrated the pre-processing required for single-cell RNA-seq analysis. kb is used to pseudoalign reads and to generate a cells x genes matrix. Following generation of a matrix, basic QC helps to assess the quality of the data.
1 2 | |
Time difference of 2.6641 mins
Feedback: please report any issues, or submit pull requests for improvements, in the Github repository where this notebook is located.